Your First GWAS Analysis
This tutorial walks you through a complete GWAS analysis from start to finish using Binx.
What You’ll Learn
- How to prepare your data for Binx
- Running a basic GWAS analysis
- Interpreting and visualizing results
- Extracting significant QTLs
Prerequisites
- Binx installed (Installation Guide)
- Genotype data (VCF or dosage format)
- Phenotype data (CSV/TSV)
Sample Data
For this tutorial, we’ll use a tetraploid potato dataset from the R/GWASpoly package.
Download the sample files:
- potato_geno.csv - Genotype data (~9,888 markers, ~1,249 samples)
- potato_pheno.csv - Phenotype data (vine maturity trait)
Citation: Rosyara, U.R., De Jong, W.S., Douches, D.S., & Endelman, J.B. (2016). Software for genome-wide association studies in autopolyploids and its application to potato. The Plant Genome 9(2).
Step 1: Examine Your Data
First, let’s look at our input files:
# Check genotype file structure
head -3 potato_geno.csv
marker,chrom,bp,AF5392-8,AF5393-1,AF5445-2,...
solcap_snp_c2_36608,chr01,508800,1,0,1,...
solcap_snp_c2_36658,chr01,527068,4,3,2,...
# Check phenotype file
head -5 potato_pheno.csv
id,vine.maturity,env
AF5033-13,4.174,Hancock15
AF5153-11,7.674,Hancock15
AF5281-4,4.174,Hancock15
...
Verify sample counts match:
# Count samples in genotype file (columns - 3)
head -1 potato_geno.csv | awk -F',' '{print NF-3, "samples"}'
# Count samples in phenotype file (lines - 1)
wc -l < potato_pheno.csv | awk '{print $1-1, "samples"}'
Step 2: Compute Kinship Matrix (Optional)
The kinship matrix captures genetic relationships. While Binx can auto-generate this using gwaspoly-rs’s set_k() function, computing it separately allows reuse across multiple traits:
binx kinship \
--geno potato_geno.csv \
--ploidy 4 \
--output kinship.tsv
Check the kinship matrix:
# View corner of matrix
head -5 kinship.tsv | cut -f1-5
Diagonal values should be approximately 1.0. Off-diagonal values represent relatedness between samples.
Step 3: Run GWAS
Now run the association analysis:
binx gwas \
--geno potato_geno.csv \
--pheno potato_pheno.csv \
--trait vine.maturity \
--kinship kinship.tsv \
--ploidy 4 \
--models additive \
--out gwas_results.csv
This will:
- Load genotypes and phenotypes
- Match samples between files
- Fit a mixed model for each marker
- Output association statistics
Understanding the Output
head gwas_results.csv
marker_id,chrom,pos,model,score,p_value,effect,n_obs,threshold
solcap_snp_c2_36608,chr01,508800,additive,0.64,0.227,0.084,1249,NA
solcap_snp_c2_36658,chr01,527068,additive,0.29,0.508,0.050,1249,NA
...
Key columns:
score: -log10 transformed p-value (higher = more significant)p_value: Probability of seeing this effect by chanceeffect: How much the trait changes per allele dosage unitn_obs: Sample size (non-missing)threshold: Significance threshold used
Step 4: Calculate Significance Threshold
Determine the significance threshold using M.eff (recommended, accounts for LD):
binx threshold \
--results gwas_results.csv \
--method m.eff \
--geno potato_geno.csv \
--ploidy 4 \
--alpha 0.05
Output:
Method: M.eff
Number of tests: 9886
Effective tests (M.eff): 6234
P-value threshold: 8.02e-06
-log10(p) threshold: 5.10
Step 5: Create Visualizations
Manhattan Plot
binx plot \
--input gwas_results.csv \
--plot-type manhattan \
--model additive \
--threshold 5.1 \
--title "Vine Maturity GWAS" \
--output manhattan.png
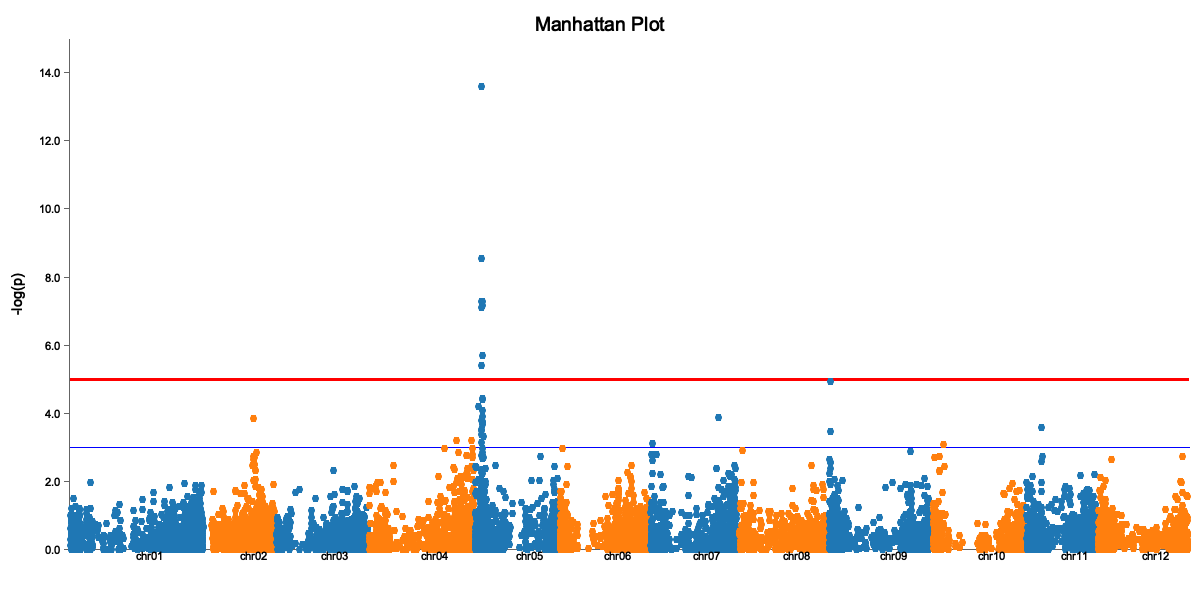
The Manhattan plot shows:
- X-axis: Genomic position (by chromosome)
- Y-axis: -log10(p-value)
- Red line: Significance threshold
- Peaks above the line are significant associations
QQ Plot
binx plot \
--input gwas_results.csv \
--plot-type qq \
--model additive \
--output qq.png
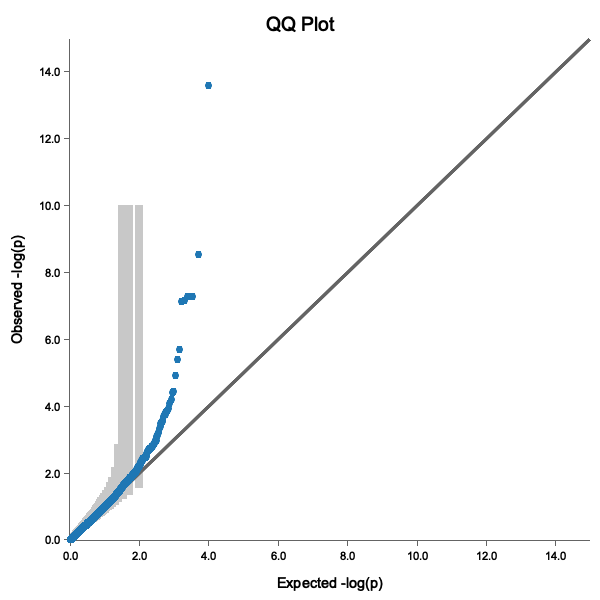
A good QQ plot shows:
- Points following the diagonal line (no inflation)
- Deviation only at the tail (true associations)
- Points within the 95% confidence band
Step 6: Extract Significant QTLs
First, run GWAS with threshold calculation to get a threshold column:
binx gwas \
--geno potato_geno.csv \
--pheno potato_pheno.csv \
--trait vine.maturity \
--kinship kinship.tsv \
--ploidy 4 \
--models additive \
--threshold m.eff \
--out gwas_results.csv
Then extract significant QTLs:
binx qtl \
--input gwas_results.csv \
--bp-window 5000000 \
--output significant_qtls.csv
cat significant_qtls.csv
marker_id,chrom,pos,model,score,effect,threshold
solcap_snp_c2_25522,chr05,4561232,additive,6.12,0.52,5.10
PotVar0067031,chr05,5193547,additive,5.89,0.48,5.10
Note: The input file must have a
thresholdcolumn. Usebinx gwas --thresholdto generate results with thresholds.
Step 7: Interpret Results
For each significant QTL:
- Effect size: A positive effect means the alternate allele increases the trait
- Position: Look up genes near the QTL position
- MAF: Very rare variants may be false positives
Candidate Gene Analysis
Once you have QTL positions, you can:
- Look up nearby genes in genome browsers
- Check if known candidate genes are in the region
- Examine the LD block around the peak marker
Complete Script
Here’s the full analysis as a script:
#!/bin/bash
set -e
# Configuration (using downloaded sample files)
GENO="potato_geno.csv"
PHENO="potato_pheno.csv"
TRAIT="vine.maturity"
PLOIDY=4
OUTDIR="results"
# Create output directory
mkdir -p $OUTDIR
# Step 1: Compute kinship
echo "Computing kinship matrix..."
binx kinship --geno $GENO --ploidy $PLOIDY --out $OUTDIR/kinship.tsv
# Step 2: Run GWAS with threshold calculation
echo "Running GWAS..."
binx gwas \
--geno $GENO \
--pheno $PHENO \
--trait $TRAIT \
--kinship $OUTDIR/kinship.tsv \
--ploidy $PLOIDY \
--models additive \
--threshold m.eff \
--out $OUTDIR/gwas_results.csv
# Step 3: Generate plots
echo "Creating plots..."
binx plot --input $OUTDIR/gwas_results.csv --plot-type manhattan --output $OUTDIR/manhattan.png
binx plot --input $OUTDIR/gwas_results.csv --plot-type qq --output $OUTDIR/qq.png
# Step 4: Extract QTLs
echo "Extracting QTLs..."
binx qtl --input $OUTDIR/gwas_results.csv --bp-window 5000000 --output $OUTDIR/qtls.csv
echo "Done! Results in $OUTDIR/"
Next Steps
- Try different genetic models
- Use LOCO for better p-value calibration
- Analyze multiple environments
- Explore polyploid-specific models
Troubleshooting
“Sample ID mismatch” error
Ensure sample IDs in phenotype file exactly match genotype column headers (case-sensitive).
Inflated QQ plot (points above diagonal)
If your QQ plot shows systematic deviation above the diagonal line:
- Try including principal components (
--n-pc 5) - Check for population structure in your data
- Use LOCO kinship (
--loco)
No significant results
- Check if trait is heritable
- Ensure sufficient sample size (>100 recommended)
- Try different genetic models